- Home
- About ACA
- Publications & Resources
- Programs
- Annual Meeting
- Membership
- -History-
- ACA Video Library
ACA Early Career Scientist Spotlight-24
Personal StatementMy fascination with structural biology took hold after first working through its entire experimental pipeline – from recombinant expression and purification to crystallization, diffraction data collection and processing. Seeing the fruits of my labor in the form of high¬-resolution electron density of the inhibitor I soaked into the crystal lattice made me appreciate the power of this experimental tool and the insights it can afford. After gaining proficiency in standard mail-in cryocrystallography, I wanted to expand my toolkit further by learning alternative X-ray based approaches to structural biology. This led me to my postdoctoral work at Cornell University where I’ve spent much of my time working on-site at CHESS (Cornell High-Energy Synchrotron Source) using the various experimental setups they offer.
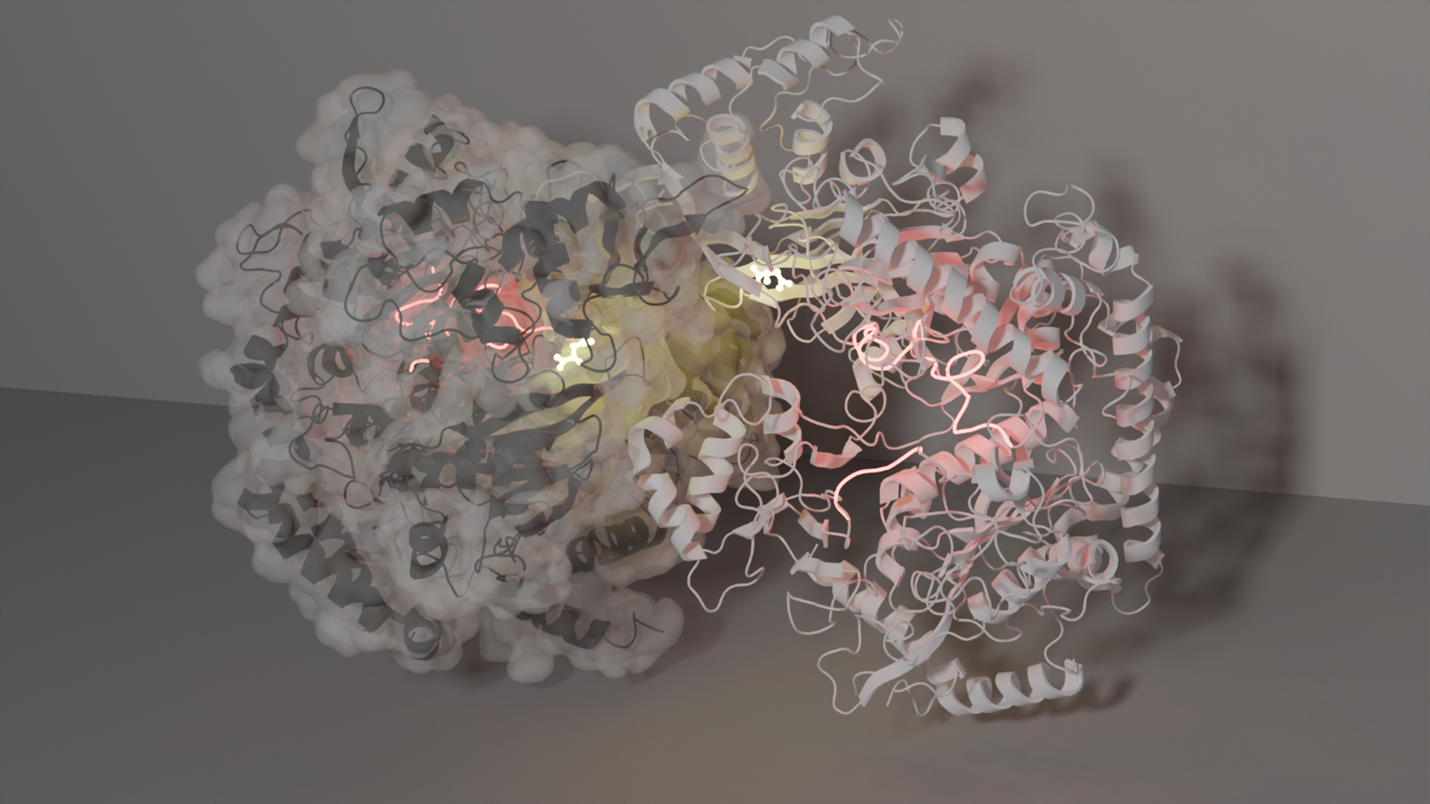
My general research interests are twofold. First, I want to gain a fundamental understanding of the biophysics of proteins/enzymes by perturbing a system of interest and viewing the structural changes. This has been the crux of my postdoctoral work, which involves using multi-temperature, time-resolved, and high-pressure X-ray crystallography. My interest in understanding the effect of temperature on enzymes traces back to my graduate studies, where I collected Arrhenius/Eyring kinetic data (enzymatic activity as a function of temperature). I wanted to understand if and how temperature changes the enzyme’s structure to achieve increased rates of turnover. Using multi-temperature X-ray crystallography, I have mapped the structural changes of a well-characterized metabolic enzyme, phosphoenolpyruvate carboxykinase (PEPCK) from -20°C to 40°C, using inhibitor mimics that faithfully recapitulate the substrate-, intermediate-, and product-bound states. This has revealed how the enzyme active site and the inhibitors/substrates along the reaction coordinate change toward more productive conformations, leading to increased turnover. A particularly surprising finding is that a dynamic lid element becomes ordered over the active site (into the productive conformation) as temperature is increased (until the enzyme and crystal begin to fall apart a high temperature). These results prompted us to re-evaluate how Arrhenius/Eyring is applied by enzymologists and others to characterize the temperature-dependent activity of enzymes. In a forthcoming publication, we argue that the assumptions made in applying these formulas are inappropriate, leading to large errors in derived thermodynamic parameters and apparent (but not actual) enthalpy-entropy compensation. Our findings should encourage biochemists to temper their interpretations of multi-temperature enzyme kinetics. Using PEPCK, we have also demonstrated a novel approach to time-resolved crystallography that we believe will greatly increase access for the broader structural biology community to time-resolved methods. Our approach involves robotically plunging crystals through a substrate film and into liquid nitrogen where the reaction is quenched. The height of the substrate film above the liquid nitrogen surface and plunge speed determines the time point sampled, and so far we have achieved 15 ms time resolution. A major advantage of our approach is that samples are prepared in the home lab and then shipped to any standard cryocrystallography beamline for remote data collection. The last biophysical axis I have been exploring is pressure, using the high-pressure capabilities at MacCHESS. For PEPCKs lid to adopt its closed, active conformation, the presence of both a nucleotide and substrate is typically required. However, with only the nucleotide present, our X-ray data show that applying a pressure of 1.5 kbar (greater than the pressure at the bottom of the Mariana trench) shifts the lid conformation to the closed state. This demonstrates the ability of pressure to reveal meaningful conformational states that might otherwise be unobservable in a given crystal form due to constraints such as the inability to soak in ligands.
My second research interest is protein evolution. By investigating how evolution creates new function, the design principles of the enzyme structure-function relationship can be revealed. Using crystallography, small angle X-ray scattering and cryoEM, I have begun studying a relatively “new” class of PEPCK, one that is unique from its two nucleotide-dependent cousins. This new class uses pyrophosphate in its catalytic mechanism and is approximately 50 kDa larger than the other classes. This extra mass, which evolution has added to the common PEPCK active site core scaffold, imparts allosteric regulation of its activity by means of oligomerization. I am now trying to map the structural transitions which lead from active monomers to inactive oligomers. In addition to describing the reaction coordinate, I hope to structurally characterize significant branches of the phylogenetic tree of this enzyme class to more comprehensively understand the influence of evolution on this scaffold. Over the last few years, my appreciation for the depth and breadth of the structural biology toolkit has grown significantly. While the methods I am using are currently “specialized”, access to them will only increase, enabling structural biologists to better understand their systems. Having been part of the ACA community for nearly a decade, I’ve learned that it is a truly supportive community that loves doing cool science. So, if you have research questions that might be best approached with these methods, reach out to those of us who have access to novel methods. We are always looking for intriguing crystals to study! **This work is in collaboration with the Thorne and Holyoak labs** |